Pyruvate Dehydrogenase Complex Deficiency (PDCD)
Zhilwan Rahim
Park University
Introduction
One of the most important molecules in the body undoubtedly is adenosine triphosphate (ATP). The process or pathways of making these molecules is considered even more important. This molecule is important to us, due to the vast requirement of it, which is involved in almost every reaction in the body. It is relatively clear the profound results that would yield in the absence and or decrease of production of this vital molecule. The process of producing ATP is similar to that of a domino effect; it consists of many metabolic pathways that ultimately lead to our product (9).
There are many different sites in this domino effect that are extremely important and necessary in order for things to run smoothly. The site, or rather complex that is going to observed, analyzed, and discussed in detail is the pyruvate dehydrogenase complex (PDC). PDC has a special job in the process of producing ATP. The conversion of pyruvate (product of glycolysis) to acetyl CoA (one of the starting products of the citric acid cycle), is accomplished by this very complex (10). As seen and discussed in great detail later on, this complex is composed of many different proteins and enzymes that work synergistically for the greater good (11).The specific site (or transition state) in which intermediate products have to be converted for the domino effect to persist is going to be the target of this investigation. The main objectives of this paper is to highlight a specific abnormality at the E1 alpha subunit that can transpire, the mechanism for this abnormality, disorders related to them, differences and similarities in Prokaryotes verse Eukaryotes, and other pathways involved.
Specific Site (PDC)
Like mentioned above, the Pyruvate dehydrogenase complex is extremely important when it comes to ATP production. Although this complex doesn’t directly yield our product, the product that is fabricated by these enzymes have a major role in the proceeding metabolic pathways that lead to ATP production (12).
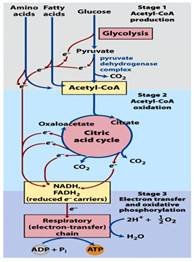
(Figure 1.1, central metabolic pathways – Lehninger Principle of Biochemistry, fifth edition)So why is PDC so important? Well, one of the biggest functions of this complex is to perform oxidative decarboxylation of Pyruvate (7). In order to fully understand the importance of this complex, a brief discussion of glycolysis is necessary.
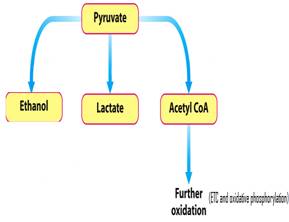
In glycolysis, one molecule of glucose (a 6 carbon molecule) is converted into two pyruvate (a 3 carbon molecule) molecules (refer to Figure 1.1). In that process (glycolysis), 2 molecules of ATP are invested to produce pyruvate, which is useless if PDC is not functioning properly.6 At this point, the pyruvate molecule has many fates. As you can see in figure 1.2, one of those paths includes the conversion of Pyruvate to ethanol or lactate (in the absence of oxygen, or abnormalities in the PDC). This occurs in a process called fermentation; the other pathway is the conversion of Pyruvate into acetyl CoA and the further oxidation of this molecule in the Citric Acid Cycle (this leads to the electron transfer chain and oxidative phosphorylation that ultimately synthesizes ATP).7,12
(Figure 1.2 Fates of Pyruvate, Biochemistry: A Short Course, 1st ed)Components and Mechanism of PDC
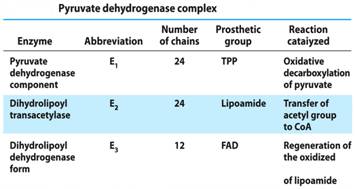
PDC is located in the mitochondrial matrix. This complex is composed of 3 enzymes: pyruvate dehydrogenase (E1), dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3).7 The complex also requires five cofactors (which are all derived from vitamins) that are vital for proper functioning of the enzymes.12 Furthermore, there are corresponding prosthetic groups that help support the PDC (refer to chart 1.3): TPP (thiamine pyrophosphate), Lipoamide, and FAD (flavin adeninedinucleotide).
(Chart 1.3 PDC. Lehninger Principle of Biochemistry, fifth edition)The step by step process of how pyruvate is converted into acetyl CoA should be discussed in detail for clarification purposes.
In step 1, pyruvate reacts with the bound thiamine pyrophosphate (TPP) of pyruvate dehydrogenase (E1), undergoing decarboxylation to the hydroxyethyl derivative (see Figure 1.4). Pyruvate dehydrogenase is also responsible for carrying out step 2, the transfer of two electrons and the acetyl group from TPP to the oxidized form of the lipoyllysyl group of the enzyme, dihydrolipoyl transacetylase (E2), to form the acetyl thioester of the reduced lipoyl group. In Step 3 we have a trans-esterification in which the SH group of CoA replaces the SH group of E2 to yield acetyl-CoA and the fully reduced (dithiol) form of the lipoyl group. In step 4 dihydrolipoyl dehydrogenase (E3) promotes the transfer of two hydrogen atoms from the reduced lipoyl groups of E2 to the FAD prosthetic group of E3, restoring the oxidized form of the lipoyllysyl group of E2. In step 5, the reduced FADH2 of E3 transfers a hydride ion to NAD+, forming NADH. The PDC is now ready for another catalytic cycle (5).
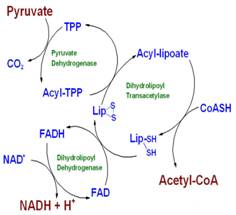
(Figure 1.4 Mechanism of PDC. Biochemistry: A Short Course, 1sted)To review of what has happened thus far, we took a glucose molecule and produced two molecules of pyruvate through a process called glycolysis. In an oxygen rich environment (and the proper functioning PDC), pyruvate is converted into acetyl CoA through a multi-step, multi-enzyme subunit complex and transferred into the mitochondria. Acetyl CoA is then put into the citric acid cycle (CAC) to propagate another pathway that ultimately leads to the production of reduced FADH+ and NADH2 (4). These electron carriers are going to run through the electron transport chain, which directs ATP synthesis. One can now see the importance of PDC and how it links the metabolic pathways together (8). If an abnormality were to occur, then the outcome would be profound.
E1 Alpha Gene Mutation
Frye and Benke1 state that “Pyruvate dehydrogenase complex deficiency is one of the most common neurodegenerative disorders associated with abnormal mitochondrial metabolism.” Looking back at the metabolism of glucose, we observed the process by which pyruvate was produced at the end of glycolysis. We then turned our attention to the many fates of pyruvate after being produced. Before mentioning those fates, the difference between Prokaryotes and Eukaryotes must be identified in regard to PDC.
When analyzing PDC in both Prokaryotes and Eukaryotes, it is rather simple because the differences are little to none. Prokaryotes have a similar cluster of proteins that resemble the PDC in eukaryotes. The only difference that has been identified seems to be the size and amount of subunits contained within the complex (11). In backtracking to the different fates of pyruvate, it can be converted into7 ethanol, lactate, and acetyl CoA molecules. In lower level eukaryotic organisms, ethanol is the pathway of choice proceeding after pyruvate production (e.g., yeast producing drinking alcohol); while in humans and many other higher level eukaryotic organisms lactate production is essential (12). Lactate production most commonly transpires under two conditions: when oxygen levels aren’t sufficient enough for the electron transport chain to take place (oxygen is the terminal electron acceptor) and when an abnormality is present in the PDC (7). Some of the byproducts of fermentation (production of lactate) are vital in humans, while too much is toxic and possibly fatal.2 The important byproducts of lactic fermentation includes the regeneration of NAD+ (electron carrier) and the elimination of accumulated pyruvate (in an anaerobic state). These two conditions are needed for glycolysis to continue (10).
What is the mechanism of abnormalities in PDCD? Thus far, we’ve discussed the fates of pyruvate when fermentation occurs. Next, the dysfunctions that occur due to a mutation in the X-linked E1 alpha gene are analyzed. We’ve established that the E1 (pyruvate dehydrogenase) enzyme is responsible for oxidative decarboxylation of pyruvate in a two-step process (5). If a mutation occurs that affects this enzyme, then pyruvate couldn’t move forward to become acetyl CoA. If acetyl CoA is not formed, then the CAC is terminated and many problems arise. The severity of PDCD depends on the percent of decreased activity level of the enzyme E1 (2). An individual with an enzyme activity level of 25 percent or higher has been found to have less severe symptoms, usually ataxia (lack of coordination of muscle movements) and psychomotor delay. Normally, infants with a 15 percent or less PDC activity level don’t survive the newborn period (2). In the case of PDCD, not only does the body have a diminished ability to produce ATP but lactic acid concentrations increase (leading to acidosis) and many cells are destroyed, especially neurological cells (1).
PDCD is considered to be a rare disease, affecting less than 200,000 Americans (but increasing steadily) annually.13 The prognosis for this disease is poor; there are no treatments with significant results at this time. One of the only treatments that provides hope for patients dealing PDCD is keeping lactic acid concentrations low in the body. This is accomplished by putting patients on a Ketogenic diet (1). This diet is similar to that of bodybuilders. It consists mostly of proteins and some fats. By consuming a diet high in proteins and fats you bypass glycolysis (glucose metabolism) and move into the CAC. Because fats and proteins do not utilize the PDC, we eliminate the accumulation of acid buildup in our cells. Unfortunately, this is not normal physiology and, therefore, not significantly helpful (1).
Conclusion
In conclusion, we learned the importance of the pyruvate dehydrogenase complex and its crucial role in the production of ATP. Initially, we discussed the importance of ATP and how every cell in the body utilizes this molecule to perform its functions (7). The different fates of pyruvate were also mentioned several times and it was figured that one path in particular was vital (Acetyl CoA production).12 Through my research, I’ve come to understand that both eukaryotes and prokaryotes share the same complex to convert pyruvate into acetyl CoA (7). Most abnormalities that result from PDCD are neuromuscular, which is due to the high ATP recruitments from these cells. The biggest factor that determines abnormality in the organism is the activity level of the enzymes involved (1). Depending on those levels, prognosis is assigned. Below 25 percent PDC activity level is considered to yield poor prognosis, and above this level one should feel good about the outcome.
The chief driving force behind constructing and researching this particular disorder is to enlighten people of the importance of understanding mitochondrial disease in general. Over the last few decades as a nation, we’ve realized how important the functioning mitochondrion is to an organism. More and more research is being conducted to understand this divine organelle in its entirety. The significance of understanding this genetic disorder is an instrumental and vital component to the whole picture. By shedding light on a rare genetic disorder such as the PDCD, the scientific community can hopefully draw parallels to other, more serious mitochondrial diseases (e.g., some forms of dementia).
References
- Baranano, K.; and Hartman, A. Current Treatment Options in Neurology: The Ketogenic Diet, 2010. Volume 10, pp. 410-419.
- Frye, R.; and Benke, P. Pyruvate Dehydrogenase Complex Deficiency.Emedicine [Online], November 6, 2009. http://emedicine.medscape.com/article/948360-overview
- Frank, R.; Pratap, J.; Pei, X.; Perham, R.; and Luisi, B. (2005). Structure 13, 1119–1130
- Johnson, P. PDCD. Health line [Online], 2002. http://www.healthline.com/galecontent/pyruvate-dehydrogenase-complex-deficiency
- Johnson, A. P. The Gale Encyclopedia of Genetic Disorders:Pyruvate dehydrogenase complex deficiency. Laurie Fundukian. 3rd ed. Detroit: Gale, 2010. Volume 2.
- Smolle, M.; and Lindsay, J. Molecular Architecture of the Pyruvate Dehydrogenase Complex: Bridging the Gap. 2006.http://www.biochemsoctrans.org/bst/034/0815/0340815.pdf
- Tymoczko, J.; Berg, J.; and Stryer, L. Biochemistry: A Short Course, 1st ed.; W.H. Freeman and Company: New York, 2010.
- The Biochemistry Questions Site. Carbohydrate Metabolism .http://biochemistryquestions.wordpress.com/2008/page/49/
- Xing, J.; Liao, J.; and Oster, G. (2005) Nat. Acad. Sci. U.S.A.Making ATP. Pp. 16539-16546
- Boiteux, A., and Hess, B. Design of Glycolysis. The Royal Society [Online], June 26, 1981, pp5-22. http://www.jstor.org/...
- Stoop, J.; Cheng, R.;Yazdi, M.; Maeug, C.;Schroeter, J.; Klueppelberg, U.; Kolodzig, S.; Baker, T.; and Reed, L. On the Unique Structural Organization of the Saccharomyces cerevisiae Pyruvate Dehydrogenase Complex. J. Biol. Chem.1997, 272, 5757-5764
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; and Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, 2002.
- Schaefer, A.; Taylor, R.; Turnbull, D.; and Chinnery, P. The Epidemiology of Mitochondrial Disorders-Past, Present and Future. Bioen. 2004, 1659, 115-120.
|