The Role of Cortisol and Abdominal Obesity in the Epidemic of Type 2 Diabetes
Tracey Liebman
University of Pennsylvania
Abstract
A rising number of individuals in the US are overweight and obese; although the morbidity and mortality rates for countless diseases have been reduced due to advances in medical research and high standard of living, the rate of type 2 diabetes mellitus increases. The culture of our affluent nation has promoted the consumption of high caloric, processed food along with physical inactivity. In the attempt to control the rise of type 2 diabetes, new treatments such as these 11β-HSD1 inhibitors and others that focus on mechanisms relating to cortisol regulation may have favorable results. Diabetes prevalence is increasing because the population is aging, people are progressively more overweight and physically inactive, and minority groups that seem more susceptible to diabetes make up an increasing percentage of the US population. Cortisol may play an important role; it is possible that even small increases in cortisol, within the range of normal, may have a detrimental influence by worsening diabetes and increasing complications. This paper is a review of the role of cortisol and abdominal obesity in the epidemic of type 2 Diabetes. As diabetes continues to grow in prevalence, the problems will become even more extensive and debilitating for society if optimal preventative measures are not taken. Future efforts in this related struggle against both obesity and type 2 diabetes should encompass a strong focus on cortisol so such prevention and treatment can successfully advance.
Diabesity: A Connection between Obesity and Diabetes
A rising number of individuals in the US are overweight and obese; although the morbidity and mortality rates for countless diseases have been reduced due to advances in medical research and high standard of living, the rate of type 2 diabetes mellitus only increases. The culture of our affluent nation has promoted the consumption of high caloric, processed food along with physical inactivity. According to the US National Commission on Diabetes, the likelihood of acquiring type 2 diabetes is 2-fold for mildly obese individuals, 5-fold for moderately obese individuals, and 10-fold for the severely obese individuals (Pi-Sunyer, 2007). The disease is costly, difficult to treat, and reduces life expectancy by 8 to 10 years (Astrup, 2000). The many complex mechanisms leading to diabetes are not yet fully understood, but the term diabesity has been suggested (Abate, 2003). Abdominal obesity has been strongly correlated with insulin resistance and type 2 diabetes, and cortisol may be involved (Franco, 2001).
Diabetes: Symptoms and Diagnosis
Type 2 Diabetes develops through abnormal insulin action and insulin secretion (Goldstein, 2002). Diabetes is diagnosed with blood glucose levels over 126mg/dL with a fasting blood glucose test and with levels over 200 mg/dL with an oral glucose tolerance test (NIH Senior Health, 2004). Many patients may not realize that they have diabetes; some may have no signs or may have only mild, seemingly common symptoms that do not appear to directly implicate diabetes. The burdens of the disease with its dangerous complications include a seriously increased need for amputations and a higher risk for heart attack, stroke, retinopathy, and nephropathy (Astrup, 2000).
Cortisol Regulation and Dysregulation: A Potentially Influential Contribution to Obesity & Diabetes
The particular mechanism of cortisol involvement in the development of obesity and diabetes is not completely clear. A direct relationship between diabetes and cortisol concentration has not been fully established, and many studies show a range of results. Circulating levels of cortisol are determined by the metabolism of cortisol, by the rate of its secretion from the adrenal cortex, and by the receptor sensitivity to cortisol (Andrews, 1999). Therefore, cortisol may be increased by various mechanisms, which include impaired metabolism, stress, and conversion of cortisone to cortisol. Although some studies have illustrated that type 2 diabetics do not have higher levels of ACTH or hypothalamic-pituitary-adrenal (HPA) axis activity than normal individuals, other studies have alternatively illustrated that cortisol levels are increased in diabetics (Chiodini, 2007). The particular techniques used, however, greatly affect the interpretation of the cortisol measurements.
Active glucocorticoids impair the activity of growth hormone, pituitary gonadotropin, and thyroid stimulating hormone. Cortisol is also involved in increasing target tissue resistance to these hormones (Chrousos, 2000). Because glucocorticoids are antagonists of insulin, they increase lipolysis and promote hepatic gluconeogenesis (Stulnig, 2004). Cortisol can stimulate caloric intake; this may contribute to obesity (Dallman, 2004). Researchers have proposed a variety of pathways that may be involved in developing excess adiposity and diabetes, and cortisol appears to be particularly essential. The most important effects of cortisol on diabetes appears to be mechanisms that involve the overall HPA axis and also complex mechanisms that implicate 11 beta-hydroxysteroid dehydrogenase type 1, adipokines, and free fatty acids with effects in adipose tissue, liver, muscle, and pancreas.Overall Cortisol Levels and HPA Axis Dysregulation
Negative feedback typically prevents excessive cortisol production, but this feedback mechanism may be hampered by factors such as emotional or physical chronic stressors, advanced age, and adverse childhood stressors (Chrousos, 2000). These stressors, or others such as smoking, alcohol, depression, or dysphoria, may contribute to abnormal HPA axis activity. If the HPA axis activity is increased, excess cortisol may be secreted (Samaras, 1997). However, in some cases plasma cortisol levels may be not appear to be altered because cortisol levels may be lowered in peripheral areas by other mechanisms (Andrews, 2002). Continuous secretion or administration of excess cortisol has been associated with depression, immunosuppression, osteoporosis, and hypertension (Chrousos, 2000). This surfeit of cortisol is also correlated with insulin resistance, visceral obesity, and dyslipidemia (Chrousos, 2000). In extreme cases of cortisol dysregulation, the clinical syndromes of Addison’s disease and Cushing’s syndrome are respectively due to cortisol deficiency and cortisol excess.
Hypothalamic-Pituitary-Adrenal Axis: Effects of Stress and Cortisol on Visceral Obesity
Stress likely affects HPA activity and increases cortisol in the body. For instance, in laboratory studies in which corticosterone, the active glucocorticoid expressed in mice detailed in Figures 1 and 2, is stimulated by stress or is externally provided, abdominal obesity has been induced in mice (Drapeau, 2003). Additionally, when an adrenalectomy is performed and glucocorticoids are therefore removed in lab experiments on animals, the development of obesity is foiled (Dallman, 2004). In a controlled, prospective study, male Cymologus monkeys under stressful conditions, which consisted of frequent social reorganization, accumulated greater intra-abdominal fat than those who were not placed under such stress (Drapeau, 2003).
Figure 1: Corticosterone (active glucocorticoid in rodents) (http---www.icgeb.org-~p450srv-ligand-corticosterone.html)
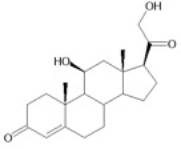
Figure 2: Glucocorticoids in Humans and Rodents
At this point, no studies in humans have been conducted that establish a direct connection between visceral adipose tissue accumulation and stress response (Drapeau, 2003). Studies have, however, illustrated that a stressful environment is often accompanied by visceral obesity. For instance, one study demonstrated that women with high waist-to-hip ratios (WHR) had significantly increased chronic work and financial stressors than those with low waist-to-hip ratios (Drapeau, 2003). More research should be conducted more specifically to determine causality of these essential relationships in humans.
Effects of Stress and Cortisol on Diabetes Complications
It is possible that even small increases in cortisol, within the range of normal, may have a detrimental influence by worsening diabetes and increasing complications. For instance, in a recent cross sectional study of 190 patients with type 2 diabetes, those with the highest cortisol levels within the range of normal had the highest levels of metabolic disturbances, as measured by fasting, postprandial and urinary glucose levels, glycosylated hemoglobin, and blood pressure. The relative abdominal mass, as estimated by sagittal diameter and body mass index (BMI), was also associated with increasing cortisol levels, but this effect was only observed when removing from the group those subjects who had lost many calories through glucosuria over 10g/l. Since this association was seen only after the data were altered, this information may be limited (Oltmanns, 2006).
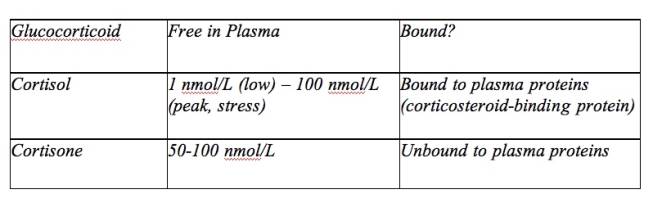
The local cortisol levels in adipose tissue may have been altered, but these were unfortunately not measured (Bjorntorp, 1999). Salivary cortisol, the measurement of choice in this study, only evaluated the unbound, active cortisol; these free cortisol concentrations were 35-50 percent lower than cortisol levels in blood. Fortunately, this is a noninvasive measure that can be conducted exactly when and where is necessary if a study requires for a timed measurement; the measure can illustrate important changes in cortisol level that occur throughout the day. It is not likely to cause stress and cortisol to increase, which is a problem with other measures. Salivary cortisol levels and free serum cortisol levels have been consistently found to be correlated. Since the measure is of only the free cortisol, which makes up less than 5 percent of plasma cortisol, even small changes within the salivary cortisol level may cause potentially important changes (Oltmanns, 2006; Rosmond, 2003; Rosmond, 2005). Because these researchers controlled the caloric intake, actions, and sleep of the subjects before and during the study, this may be more informative than studies on outpatients that have had less control over confounding factors (Oltmanns, 2006). Additionally, the 190 subjects is a relatively large sample size that may provide valuable data. This study, among many others, does not determine causality and does not illustrate any long-term data.
Cortisol may lead to more chronic complications of diabetes and worsening levels of insulin sensitivity, but only correlation and not causation has been demonstrated. In a recent study on cortisol performed by Chiodini et al on 170 type 2 diabetic subjects and 70 non-diabetic subjects, the diabetics were separated by those with chronic complications and those without complications. Individuals suspected to have hypercortisolism were not included, as these subjects all had cortisol levels in the normal range; this likely helped the study to more effectively compare the groups equally. Subjects with severe complications, which could alter the results, were excluded. The relatively large sample size helped to limit some problems with the study, and cortisol was measured in a range of ways including 24 hour UFC, plasma ACTH levels, and serum cortisol levels, which may have helped to reduce the limitations of the various measurements.
In the diabetic group with complications, the investigators found that serum cortisol levels were associated with the number of complications; no other measurement techniques, however, were correlated with complications. Because ACTH does not seem to be changed in order to respond to the change in cortisol concentration, this may signify that diabetics with complications have abnormal HPA activity and regulation (Chiodini, 2007). In diabetics with a lack of complications, HPA activity was found to be similar to that of nondiabetics. Therefore, the cortisol secretion was found to be correlated with increased complications in diabetics. Cortisol may be involved in detrimental effects on metabolic control and therefore contribute to a rise in insulin resistance (Chiodini, 2007). These damaging mechanisms are often associated with a worsening of diabetes that often is accompanied by these chronic complications.
Many other studies did not separate the subjects into groups with chronic complications and without complications, which may have been a factor in the wide array of cortisol findings in diabetics. Other studies have been inadequate because they did not separate individuals by categories of obesity, gender, or degree of metabolic control; some laboratory and murine studies cannot be generalized to human clinical situations. It is also difficult to properly determine the source of cortisol abnormalities, which may include issues with cortisol secretion, cortisol metabolism, or receptor sensitivity (Andrews, 2002). Although the glucocorticoid receptor expression may play a role, this is also challenging to determine in humans.
Cortisol in Adipose Tissue: Visceral vs Subcutaneous Fat, 11β-HSD1, and Adipokines
Measuring Obesity: The importance of visceral versus subcutaneous adiposity
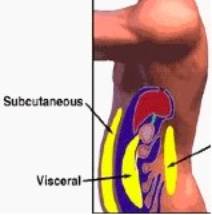
Abdominal adiposity, a strong predictor of type 2 diabetes, may be an indication of dysfunctional adipose tissue. Abdominal adiposity is often accompanied by metabolic disturbances, including insulin resistance, hypertriglyceridemia, hyperinsulinemia, glucose intolerance, hypertension, reduced levels of HDL, increased small and dense LDL, glucose intolerance, and hypertension (Carr, 2004; Despres, 2006). Several clinical features of abdominal obesity also appear in the hypercortisolism in Cushing’s Syndrome (Pasquali, 2000).
Regional differences in adipocytes are apparent and present an important issue of varying risks depending on fat type; visceral adipocytes and subcutaneous adipocytes differ in ways other than solely location. The adipocytes are crucial secretory cells; the adipocytes can affect many pathways by altering the amount of substances secreted, and adipocytes themselves have receptors and can be affected by substances such as cortisol. In a study on Japanese-American men, it was found that intra-abdominal adipose tissue was strongly associated with type 2 diabetes. In contrast, subcutaneous adipose tissue in the thigh, thorax, and abdomen, pictured in Figure 8, were not significantly correlated with diabetes (Pi-Sunyer, 2006). Additionally, in a study of monozygotic twins, those twins with greater differences in visceral adipose tissue also exhibited greater disparities in insulin sensitivity (Bonora, 2000). Higher levels of visceral fat have been correlated with excess hepatic glucose production and lower levels of glucose removal (Bonora, 2000). Even if overall weight is unchanged, there is a greater risk for diabetes with a relative increase in visceral abdominal fat. An inverse correlation has been observed between visceral abdominal fat and insulin sensitivity in both nonobese and obese individuals (Bonora, 2000). Therefore, normal weight individuals may not realize they could be at risk for diabetes, especially with clinical assessments currently used to determine risk that only consider overall weight and do not account for abdominal adiposity.Figure 3: Visceral and Subcutaneous Adipose Tissue (www.arfamilies.org)
A variety of tools have been used to measure obesity, but techniques that particularly assess abdominal obesity may more effectively reveal risk than those that only evaluate overall obesity. Abdominal obesity is often not illustrated properly when BMI or weight are used. Furthermore, these measurements may be population specific, and it may not be appropriate to use the same cut-off values for all races and ethnicities (Samaras, 1997).
Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) seem to be the optimal risk assessment tools to measure adiposity, but both have high costs and are not often used (Samaras, 1997). CT is beneficial because it can distinguish between intra-abdominal and subcutaneous abdominal fat. This differentiation may help researchers more specifically identify risk factors and mechanisms of disease progression (Samaras, 1997). The waist to hip ratio (WHR) is the most common anthropometric measure used; yet, WHR does not separate the measurement of visceral and subcutaneous abdominal compartments (Stulnig, 2004; Franco, 2001; Gautier, 1998). Therefore, it may not serve as the optimal evaluation tool. However, some studies assert that WHR can assess risk as well as WC (waist circumference) can (Bonora, 2000). The sagittal abdominal diameter (SAD), which measures the distance between the anterior wall of the abdomen and back, and WC)and may be more effective tools for evaluating risk. WC and waist-height may have a closer relationship with intra-abdominal fat content than WHR does, as shown by CT (Samaras, 1997). WC also seems to be particularly associated with insulin resistance, but WC cannot evaluate the ratio of visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT) (Misra, 2003; Bonora, 2000).
As an example, in a case control study by Anjana et al with 82 diabetic individuals and 82 matched nondiabetics, there was a strong association of visceral and central abdominal fat with type 2 diabetes, which was similarly illustrated by WC and SAD (Anjana, 2004). When using these measurement tools, subcutaneous abdominal fat was not associated with diabetes. Dual-energy x-ray absorptiometry, known as DEXA, is useful to use with a large population, but it measures total and central abdominal fat without distinguishing between VAT and SAT (Samaras, 1997). In the Anjana study, however, because central abdominal fat measured by DEXA was strongly associated with the visceral fat measured by CT, DEXA may serve as a useful tool (Anjana, 2004). Future patients and research studies will benefit from the determination of the most effective tools, which will lead to optimal identification of disease risks and proper focus on finding fundamental causative mechanisms.Adipose Tissue: Cortisol and 11β-HSD1
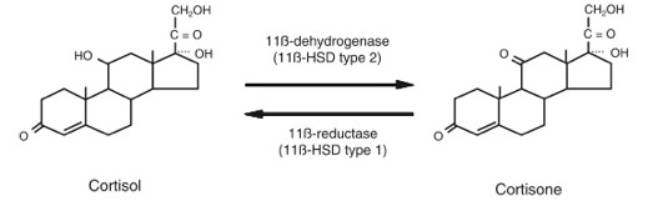
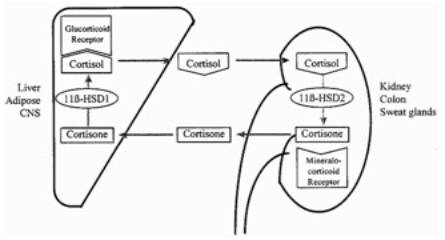
In adipose tissue, a crucial feature seems to be 11 beta-hydroxysteroid dehydrogenase type 1, known as 11β-HSD1, shown in Figure 3. Cortisone, an inactive glucocorticoid, is converted to cortisol, an active glucocorticoid, by this enzyme that exhibits reductase activity. A rise in 11β-HSD1 action, and therefore cortisol activity, may have various effects that are still under investigation. In adipose tissue, cortisol may promote differentiation and proliferation of adipocytes (Stulnig, 2004). Other than in adipose tissue, 11β-HSD1 may act in the liver, muscle, brain, and other organs. Using a stable isotope tracer in the body to isolate how much cortisol is converted from cortisone, the amount of cortisol found to be produced in this conversion is approximately 25 percent of the amount produced by the adrenal cortex (Walker, 2006).
Figure 4: 11β-HSD1 Action- Conversion between Cortisone and Cortisol (Walker, 2006)
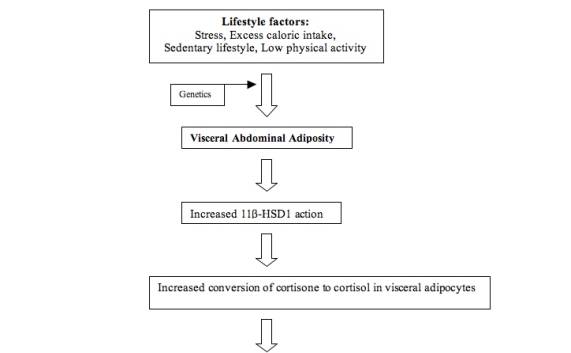
Adipose tissue is exposed to an increased cortisol concentration when there is a rise in 11β-HSD1 action (Stulnig, 2004). There are more glucocorticoid receptors in visceral adipocytes compared to subcutaneous adipocytes, so 11β-HSD1 locally converts more cortisone to cortisol in visceral adipocytes (Dallman, 2004; Franco, 2001). Because there is a constant supply of cortisone in the plasma, as shown in Figure 7, well-regulated target tissues may adjust cortisol concentrations through this conversion mechanism. Under some conditions in vitro, 11β-HSD1 may convert cortisol to cortisone, but the extent of this dehydrogenase activity in the body is unknown (Andrew, 2005). In a study of transgenic mice engineered to overexpress 11β-HSD1, there was an increase in adipose levels of corticosterone, even though circulating glucocorticoids remained at normal levels (Masuzaki, 2001). In another part of the body, such as the kidney, there may be a rise in activity of 11β-HSD2. This different isoform of 11β-HSD converts active glucocorticoids to inactive glucocorticoids, as shown in Figure 4 (Seckl, 2001). This allows aldosterone, rather than an active glucocorticoid, to access mineralocorticoid receptors (Qi, 2007).
Figure 5: Cortisone and Cortisol in Plasma (Seckl, 2001)
Figure 6: Conversions between Cortisone and Cortisol (Seckl, 2001)
A case control study performed by Andrews et al did address the issue of abnormal, varied cortisol concentrations in diabetic men of normal weight (Andrews, 2002). Cortisol levels were evaluated by several methods; plasma and urine samples were taken, and 11β-HSD1 activity was measured by taking a biopsy of subcutaneous fat. No total cortisol levels were found to be different between diabetics and controls by the measures of fasting morning plasma cortisol and total urinary cortisol metabolite excretion rate, which reflect HPA axis activity. Subcutaneous adipose tissue 11β-HSD1 activity was unchanged, but hepatic 11β-HSD1 activity was decreased in diabetics.
This study appropriately controlled for many variables often not addressed in other studies with less adequate methodologies; this study included only men to control for gender and did not include patients with obesity or hypertension so these confounding factors would not alter results. The chosen subjects did not exhibit any complications of diabetes, which is advantageous because many studies are problematic due to the fact that those with and without complications are grouped together.
Andrews et al also used the sum of urinary metabolites of cortisol in urine over 24 hours. This is considered a superior measure to urinary free cortisol (UFC). The UFC estimates the level of average circulating free cortisol, but this represents less than 5 percent of the total cortisol metabolite excretion. Additionally, it may be difficult to perform measurements of the UFC at an exact time, if the levels are supposed to be taken at specific intervals, because the investigator must rely on the subject to urinate on schedule (Bjorntorp, 1999).
Another measure that was not used in this study, the urinary ratio of cortisol to cortisone metabolites, provides information about the conversion between cortisol and cortisone; the potentially important findings of increased or decreased 11β-HSD1 activity in different compartments would not be revealed in this method. Unfortunately it would be difficult to measure the activity in visceral abdominal fatthat surrounds crucial organs, so this was not taken into account. It may have also been more favorable to have a sample size greater than 25 diabetics and 25 controls (Andrews, 2002).
Adipose Tissue: Cortisol and Adipokines
Glucocorticoids influence the release of adipokines from adipocytes; cortisol typically promotes an increase in leptin and resistin but decreases adiponectin (Qi, 2007). In large adipocytes present in obesity, these changes are often observed. Adipose cells secrete peptide and non-peptide substances; therefore, the adipocytes can communicate with other cells. For instance, adipocytes secrete resistin, and increasing levels of this protein are often associated with obesity; this is one potential link between adiposity and insulin resistance. (Franco, 2001). This adipokine action is likely to escalate insulin resistance (Pi-Sunyer, 2007). Larger visceral adipose cells additionally have greater secretion of TNF-a, which leads to insulin resistance in rodents and IL-6 that amplifies the inflammatory response (Pi-Sunyer, 2007).
Cortisol in Liver
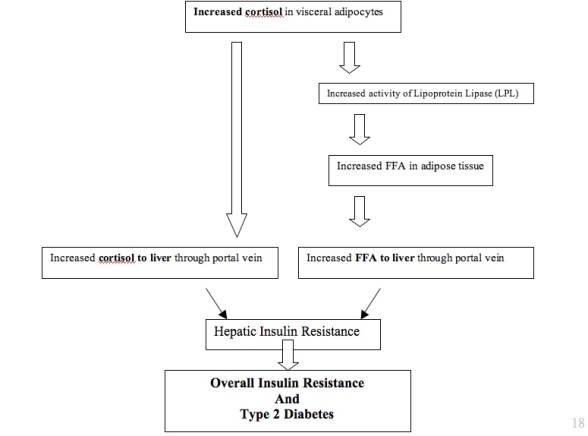
Because the liver is a major target for glucocorticoids, the effects on the liver leading to insulin resistance likely affect the entire body. This is likely because the liver is responsible for most of the glucose overproduction involved in type 2 diabetes (Bergman, 2000). The measurements of glucocorticoid metabolism for the body as a whole usually reflect the activity in the liver (Tomlinson, 2005). The high levels of glucocorticoids in visceral obesity increase the activity of lipoprotein lipase (LPL), so lipoprotein triglycerides are converted to free fatty acids. Subcutaneous adipocytes are more sensitive to insulin, but visceral adipocytes are more sensitive to glucocorticoid stimulation. Therefore, lipolysis of fat, inhibited by insulin, is more pronounced in visceral fat than in subcutaneous fat; this pathway involving cortisol in visceral adipose tissue and the liver is detailed in Figure 5 (Goldstein, 2002). This increases the release of free fatty acids (FFA) in visceral adipose tissue (VAT); because VAT drains into the portal vein, this may increase the free fatty acids, glucocorticoids, and adipokines that reach the liver through the blood and contribute to hepatic insulin resistance (Samaras, 1997; Rosmond, 2003). In contrast, subcutaneous adipose tissue (SAT) drains free fatty acids into systemic circulation so the liver would not be directly affected (Bonora, 2000).
Figure 7: Proposed Pathogenesis of cortisol involvement in pathway through abdominal adiposity and the liver to insulin resistance.
In the liver, the free fatty acids promote gluconeogenesis and inhibit the suppression of glycogenolysis. Specifically, the increased glucocorticoids promote expression of phosphoenolpyruvate carboxykinase (PEPCK) and upregulate the glucose-6-phosphatase system (G-6-Pase), which increases the production of glucose. The glucose overproduction contributes to hepatic insulin resistance and eventually insulin resistance throughout the body (Qi, 2006; Bergman, 2000). In obese individuals, though 11β-HSD1 expression is typically increased in adipose tissue, many studies show that its expression is usually decreased in the liver (Stulnig, 2004; Tomlinson, 2005). Direct cortisol measurements in the portal vein, however, are difficult to perform effectively; it has only been previously done during surgical procedures, and the effect of the surgery on the body may distort the data (Andrew, 2005).
Cortisol in Muscle and Pancreas
In myoblasts, increased expression of 11β-HSD1 and the glucocorticoid receptor (GR) in muscle are correlated with increased insulin resistance and BMI (Stulnig, 2004). In insulin resistant patients, there may be a rise in number of muscle glucocorticoid receptors. Because visceral adiposity involves the collection of fat around the organs, adipocytes may accumulate in muscles. The high glucocorticoid levels inhibit glycogen synthase and promote the release of free fatty acids. Additionally, the glucocorticoids in muscles may promote the expression of fatty acid transporter genes (Qi, 2007). Therefore, triglycerides may collect in the muscles, and muscles may be affected with hyperinsulinemia (Rosmond, 2003; Abate, 2003). Glucocorticoids likely act on downstream insulin signaling in the muscle and therefore decrease the uptake of glucose, contributing to insulin resistance and type 2 diabetes (Qi, 2007). In the pancreas, cortisol can impair insulin-dependent uptake of glucose. Also, increased cortisol produced by increased 11β-HSD1 action in islet cells may hinder the secretion of insulin from beta cells of the pancreas (Stulnig, 2004; Rosmond, 2005).
Risk Factors for Developing Abdominal Obesity and Diabetes
The development of abdominal obesity is affected by environmental, hormonal, and genetic factors. However, the importance of potential influences on preferential regional accumulation of body fat distribution have not been elucidated (Franco, 2001). Socioeconomics status (SES) was correlated with BMI in the NHANES III study. In developed countries, individuals often have an inactive lifestyle and eat high calorie-dense foods (Despres, 2006). The differences in diabetes prevalence between ethnicities may be a result of such socioeconomic factors (Abate, 2003).
In individuals who are overweight or normal weight, intra-abdominal fat increases with age, despite other changes in total body fat (Carr, 2004). The highest prevalence in diabetes is presently in people over 65 years old. Aging-associated changes in body fat distribution may intensify diabetes risk (Goodpaster, 2003). Smoking may advance insulin resistance by reducing blood flow to muscles, raising sympathetic nervous system activity, increasing lipolysis, and increasing free fatty acids (Samaras, 1997).Reducing Risks: Prevention of Diabetes and Future Trends in Diabetes, Current Strategies
Proactive prevention and treatment of overweight and obesity is crucial in order to effectively combat type 2 diabetes. In 2000, 171 million people had diabetes worldwide, and 366 million people are anticipated to have diabetes by 2030 (Chaturvedi, 2007). Diabetes cost the US $132 billion in 2002; this includes $92 billion of direct costs and $40 billion of indirect costs (NIDDK, 2006). Between 1987 and 2002, the population death rate attributed to diabetes increased 45 percent (Chaturvedi, 2007).
There is a possibility to intervene to modify the non-genetic factors involved in advancing abdominal adiposity. The most effective intervention appears to be disease prevention through lifestyle modifications of increased physical activity and improved food intake (Chaturvedi, 2007). According to a randomized study from the Diabetes Prevention Program launched in 1996 by NIDDK, individuals at high risk for developing diabetes most effectively reduced their risk, by 58 percent, of progressing to diabetes by engaging in lifestyle modifications. These changes included improved diet and 30 minutes of exercise per day; patients also lost 5 to 7 percent of their body weight. In contrast, the insulin sensitizer drug metformin was less successful, as it reduced diabetes risk less considerably, by 31 percent, as shown in Figure 6 (NIDDK, 2006). Stress management may also be beneficial, and cognitive behavioral therapy could be helpful to maintain changes (Drapeau, 2003). Further intervention for abdominal adiposity through surgery for severe cases may be advantageous in some patients (Franco, 2001).
Figure 8: Diabetes Prevention Studies (Abate, 2003)
Studies on prevention of diabetes Study Patient Population Intervention Risk Reduction Finnish DPP
(Tuomilehto et al., 2001)IGT Diet and exercise 58% US DPP
(Diabetes Prevention Program
Research Group, 2002)IGT Diet, exercise metformin 58%, 31% TRIPOD (Azem et al., 1998) Previous GDM Troglitazone 56% Future Strategies: 11β-HSD1 inhibitors
Although many other pathways and factors are involved in the development of type 2 diabetes, cortisol may actually prove to be more important than previously considered. Specifically, the use of 11β-HSD1 inhibitors may be able to affect the progression to obesity and diabetes. Future studies should focus more on the efficacy of these 11β-HSD1 inhibitors in humans (Tomlinson, 2005). At this point, human studies have been conducted with nonselective 11β-HSD1 inhibitors, such as carbenoloxone (Tomlinson, 2005). Although the nonselective inhibitors tend to decrease overall cortisol and decrease production of glucose, they decrease both dehydrogenase and reductase activity of 11β-HSD1. Because the nonselective inhibitors have also been shown to increase blood pressure and increase mineralocorticoids, they are not appropriate for treatment of obese and diabetic individuals (Tomlinson, 2005). Although the clinical trials with nonselective inhibitors are an important first step in investigating the effects in humans, more extensive studies must be conducted with selective inhibitors.
Selective 11β-HSD1 inhibitors, which affect 11β-HSD1 to a significantly greater extent than they affect 11β-HSD2, may be a valuable treatment for diabetic patients. These inhibitors may reduce obesity, enhance metabolic control, and decrease tissue specific production of cortisol, as shown in recent rodent studies (Stulnig, 2004; Tomlinson, 2005). In type 2 diabetic mice, certain selective 11β-HSD1 inhibitors have been shown to decrease blood glucose levels and improve insulin sensitivity.
There is, however, a significant amount of variation between species in terms of efficacy of 11β-HSD1 inhibitors. The important differences between human and rodent physiology may lead to difficulties in extrapolating data from mice to humans. For instance, mouse 11β-HSD1 was found to more efficiently perform the conversion than human 11β-HSD1. Other animals such as dogs, rats, hamsters, and guinea pigs have also been investigated, but it will not be easy to find the optimal selective 11β-HSD1 inhibitor for humans because different inhibitors seem to greatly vary in potency between species (Arampatzis, 2005). The effectiveness will also depend on the ability of the inhibitors to be targeted to certain tissues, such as adipose rather than the liver, in order to successfully prevent the excess conversion of cortisone to cortisol. Because 11β-HSD1 is present in other locations in the body, careful assessments must be performed in order to ensure that other mechanisms are not adversely affected.
In the attempt to control the rise of type 2 diabetes, new treatments such as these 11β-HSD1 inhibitors and others that focus on mechanisms relating to cortisol regulation may have favorable results. Diabetes prevalence is increasing because the population is aging, people are progressively more overweight and physically inactive, and minority groups that seem more susceptible to diabetes make up an increasing percentage of the US population. As diabetes continues to grow in prevalence, the problems will become even more extensive and debilitating for society if optimal preventative measures are not taken. Future efforts in this related struggle against both obesity and type 2 diabetes should encompass a strong focus on cortisol so such prevention and treatment can successfully advance.
References
Abate, Nicola and Manisha Chandalia. “The Impact of Ethnicity on Type 2 Diabetes.” Journal of Diabetes and Its Complications 17 (2003) 39-58.
Andrew, Ruth et al. “The Contribution of Visceral Adipose Tissue to Splanchnic Cortisol Production in Healthy Humans.” Diabetes (2005) 54(5):1364-70.
Andrews, Robert C and Brian R Walker. “Glucocorticoids and Insulin Resistance: old hormones, new targets.” Clinical Science (1999) 96.
Andrews, Robert C et al. “Abnormal Cortisol Metabolism and Tissue Sensitivity to Cortisol in Patients with Glucose Tolerance.” Journal of Clinical Endocrinology and Metabolism 87 (12): 5587-5593.
Arampatzis, Spyridon et al. “Comparative enzymology of 11b-hydroxysteroid dehydrogenase type 1 from six species.” Journal of Molecular Endocrinology (2005) 35, 89-101.
Astrup, A and N Finer. “Redefining Type 2 Diabetes: ‘Diabesity’ or ‘Obesity Dependent Diabetes Mellitus’?” The International Assoc for the Study of Obesity. Obesity Reviews 1 (2000) 57-59.
Bergman, Richard N, Ader M. “Free fatty acids and Pathogenesis of Type 2 Diabetes Mellitus.” TEM Vol 11, No 9, 2000.
Bjorntorp P, Holm G, Rosmond R. “Hypothalamic arousal, insulin resistance, and Type 2 diabetes mellitus. Diabetic Medicine (1999) 16, 373-383.
Boden, G. “Effects of Free Fatty Acids (FFA) on Glucose Metabolism: Significance for Insulin Resistance and Type 2 Diabetes.” Exp Clin Endocrinol Diabetes 2003;111:121-124.
Bonora, E. “Relationship between regional fat distribution and insulin resistance.” International Journal of Obesity (2000) 24 Suppl 2, S32-S35.
Carr, MC, Brunzell JD. “Abdominal Obesity and Dyslipidemia in the Metabolic Syndrome: Importance of Type 2 Diabetes and Familial Combined Hyperlipidemia in Coronary Artery Disease Risk.” J Clin Endocrinology and Metabolism 89(6):2601-2607.
Chaturvedi, Nish. “The burden of diabetes and its complications: Trends and implications for intervention.” Diabetes Research and Clinical Practice. 2007 May;76 Suppl 1:S3-12.
Chiodini I, Adda G, Scillitani A, Coletti F, Morelli V, Di Lembo S, Epaminonda P, Masserini B, Beck-Peccoz P, Orsi E, Ambrosi B, Arosio M. “Cortisol Secretion in Patients with Type 2 Diabetes: Relationship with Chronic Complications” Diabetes Care (2007) 30:83-88.
Chrousos, G. “The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: neuro-endocrine and target tissue-related causes.” International Journal of Obesity (2000) 24, Suppl 2, S50-55.
Dallman MF, la Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF. “Minireview: Glucocorticoids – Food Intake, Abdominal Obesity, and Wealthy Nations in 2004.” Endocrinology 145(6):2633–2638. 2004.
DeFronzo, Ralph A. “Pathogenesis of type 2 diabetes mellitus.” Med Clin N Am 88 (2004) 787-835.
Drapeau V, Therrien F, Richard D, Tremblay A. “Is Visceral Obesity a Physiological Adaptation to Stress?” Panminerva Med 2003;45:189-95.
Franco C. “Visceral Obesity and the role of the somatotropic axis in the development of metabolic complications” Growth Hormone & IGF Research 2001, Supplement A, S97-S102.
Gautier JF, Mourier A, de Kerviler E, Tarentola A, Bigard AX, Villette JM, Guezennec CY, Cathelineau G. “Evaluation of Abdominal Fat Distribution in Noninsulin-Dependent Diabetes Mellitus: Relationship to Insulin Resistance.” J Clin Endocrinol Metab 83: 1306-1311, 1998.
Gerich JE. “Is Insulin resistance the principal cause of type 2 diabetes?” Diabetes, Obesity and Metabolism, (1999)1: 257-263.
Goldstein BJ. “Insulin Resistance as the Core Defect in Type 2 Diabetes Mellitus.” American Journal of Cardiology. Vol 90 (5A) 5 September 2002.
Goodpaster BH, Krishnaswami S, Resnick H, Kelley DE, Haggerty C, Harris TB, Schwartz AV, Kritchevsky S, Newman AB. “Association Between Regional Adipose Tissue Distribution and Both Type 2 Diabetes and Impaired Glucose Tolerance in Elderly Men and Women.” Diabetes Care (2003) 26: 2.
Lazar MA. “How Obesity Causes Diabetes: Not a Tall Tale.” Science. 373 (2005); 307.
Lindmark S, Lönn L, Wiklund U, Tufvesson M, Olsson T, Eriksson JW. “Dysregulation of the Autonomic Nervous System Can Be A Link between Visceral Adiposity and Insulin Resistance.” Obesity Research (2005) 13:4.
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. “A Transgenic Model of Visceral Obesity and the Metabolic Syndrome.” Science. 2166 (2001); 294.
Misra A, Vikram N. “Clinical and Pathophysiological Consequences of Abdominal Adiposity and Abdominal Adipose Tissue Depots.” Nutrition (2003) 19:457-466.
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), "National Diabetes Information Clearinghouse." Dec 2006. 1 Mar 2007 <http://www.diabetes.niddk.nih.gov/dm/pubs/riskfortype2/index.htm>.
Oltmanns KM, Dodt B, Schultes B, Raspe HH, Schweiger U, Born J, Fehm HL, Peters A. “Cortisol correlates with metabolic disturbances in a population study of type 2 diabetic patients.” European Journal of Endocrinology (2006) 154: 325-331.
Pasquali R, Vicennati V. “Activity of Hypothalamic-Pituitary-Axis in different obesity phenotypes.” International Journal of Obesity (2000) 24, Suppl 2, S47-S49.
Pi-Sunyer, F Xavier. “How effective are lifestyle changes in the prevention of Type 2 Diabetes Mellitus?” Nutrition Reviews (2007) Vol 65. No. 3.
Qi D, Rodrigues B. “Glucocorticoids produce whole body insulin resistance with changes in cardiac metabolism.” Am J Physio Endocrinol Metab 292: 654-667, 2007.
Rosmond R. “Role of Stress in the pathogenesis of the metabolic syndrome.” (2004) Psychoneuroendocrinology (2005) 30, 1-10.
Rosmond R. “Stress Induced Disturbances of the HPA Axis: a pathway to Type 2 Diabetes?” Med Sci Monit, 2003; 9(2): RA35-39.
Samaras K, Campbell LV. “The non-genetic determinants of central adiposity.” International Journal of Obesity (1997) 21, 839-845.
Stulnig TM, Waldhausl W. “11beta-hydroxysteroid dehydrogenase Type 1 in obesity and Type 2 diabetes.” Diabetologia (2004) 47:1-11.
Tomlinson JW, Stewart PM. “Mechanisms of Disease: selective inhibition of 11β-hydroxysteroid dehydrogenase type 1 as a novel treatment for the metabolic syndrome.” Nature Clinical Practice: Endocrinology and Metabolism. (2005) 1: 92-99
Walker BR. “Cortisol – cause and cure for metabolic syndrome?” Diabetic Medicine (2006) 23: 1281-1288.

|